 16s rDNA测序首先需要提取环境样品
16s rDNA测序首先需要提取环境样品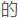 DNA
DNA 这些DNA可以来自土壤、粪便、空气或水体等任何来源。
这些DNA可以来自土壤、粪便、空气或水体等任何来源。
 提取DNA后需要经过质检和纯化
提取DNA后需要经过质检和纯化 一般16s rDNA测序扩增对DNA
一般16s rDNA测序扩增对DNA 总量要求并不高
总量要求并不高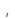 总量大于100ng
总量大于100ng 浓度大于10ng/ul一般都可以满足要求。如果是来自和寄主共生
浓度大于10ng/ul一般都可以满足要求。如果是来自和寄主共生 环境如昆虫
环境如昆虫 肠道微生物
肠道微生物 提取时可能包括了寄主本身
提取时可能包括了寄主本身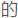 大量DNA
大量DNA 对DNA
对DNA 总量要求会提高。微生物菌群多样性测序受DNA提取和扩增影响很大
总量要求会提高。微生物菌群多样性测序受DNA提取和扩增影响很大 不同
不同 扩增区段和扩增引物甚至PCR循环数
扩增区段和扩增引物甚至PCR循环数 差异都会对结果有所影响。因而建议同一项目不同样品
差异都会对结果有所影响。因而建议同一项目不同样品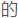 都采用相同
都采用相同 条件和测序方法
条件和测序方法 这样相互之间才存在可比性。
这样相互之间才存在可比性。
 完成PCR之后
完成PCR之后 产物一般可以直接上测序仪测序
产物一般可以直接上测序仪测序 在上机测序前我们需要对所有样本进行定量和均一化
在上机测序前我们需要对所有样本进行定量和均一化 通常要进行荧光定量PCR。完成定量
通常要进行荧光定量PCR。完成定量 样品混合后就可以上机测序。
样品混合后就可以上机测序。
 16s rDNA测序目前可以采用多种不同
16s rDNA测序目前可以采用多种不同 测序仪进行测序
测序仪进行测序 包括罗氏
包括罗氏 454
454 Illumina
Illumina Novoseq, MiSeq
Novoseq, MiSeq Hiseq
Hiseq Life
Life PGM 或 Pacbio 以及 nanopore
PGM 或 Pacbio 以及 nanopore  三代测序仪。不同
三代测序仪。不同 仪器各有优缺点
仪器各有优缺点 目前最主流
目前最主流 是Illumina公司
是Illumina公司 MiSeq
MiSeq 因为其在通量、长度和价格三者之间最为平衡。MiSeq 测序仪可以产生 2x300 bp
因为其在通量、长度和价格三者之间最为平衡。MiSeq 测序仪可以产生 2x300 bp  测序读长
测序读长 Hiseq 和 Novoseq 可以生成 2x250bp 或者 2x150bp
Hiseq 和 Novoseq 可以生成 2x250bp 或者 2x150bp  测序读长
测序读长 且通量较大。
且通量较大。
 16s rDNA分析基本流程:
16s rDNA分析基本流程:
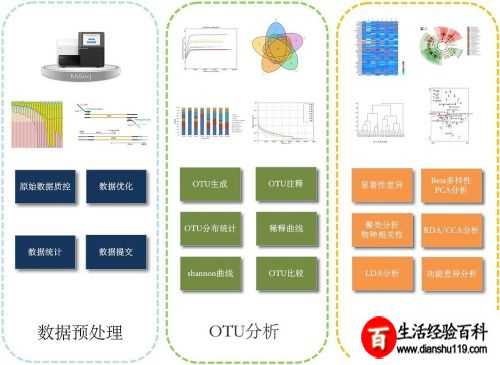
 原始数据处理:
原始数据处理:
原始测序数据需要去除接头序列
 根据 overlap 并将双端测序序列进行拼接成单条序列
根据 overlap 并将双端测序序列进行拼接成单条序列 同时对序列质量进行质控和过滤。提供已知数据库 GreenGenes 作为参考
同时对序列质量进行质控和过滤。提供已知数据库 GreenGenes 作为参考 去除嵌合体序列得到最终可用
去除嵌合体序列得到最终可用 序列。
序列。
提取出
 数据以 fastq 格式保存
数据以 fastq 格式保存 每个样本有 fq1 和 fq2两个文件
每个样本有 fq1 和 fq2两个文件 里面为测序两端
里面为测序两端 reads
reads 序列按顺序一一对应。
序列按顺序一一对应。
原始fastq格式是一个文本格式用于存储生物序列(通常是核酸序列)和其测序对应
 质量值。这些序列以及质量信息用ASCII字符标识。
质量值。这些序列以及质量信息用ASCII字符标识。
 OTU分类和统计:
OTU分类和统计:
OTU(operational taxonomic units) 是在系统发生学研究或群体遗传学研究中
 为了便于进行分析
为了便于进行分析 人为给某一个分类单元(品系
人为给某一个分类单元(品系 种
种 属
属 分组等)设置
分组等)设置 同一标志。通常按照 97%
同一标志。通常按照 97%  相似性阈值将序列划分为不同
相似性阈值将序列划分为不同 OTU
OTU 每一个 OTU 通常被视为一个微生物物种。相似性小于97%就可以认为属于不同
每一个 OTU 通常被视为一个微生物物种。相似性小于97%就可以认为属于不同 种相似性小于93%-95%可以认为属于不同属。样品中微生物多样性和不同微生物丰度都是基于对OTU分析。
种相似性小于93%-95%可以认为属于不同属。样品中微生物多样性和不同微生物丰度都是基于对OTU分析。
使用QIIME(version 1.8.0)工具包进行统计注释。

使用QIIME(version 1.9.0, http://bio.cug.edu.cn/qiime/)ucluster方法根据97%序列相似度将所有序列进行同源比对并聚类成operational taxonomic units (OTUs)。然后与数据库GreenGenes(version gg_13_8, http://greengenes.lbl.gov/cgi-bin/JD_Tutorial/nph-16S.cgi)进行比对比对方法uclustidentity 0.9 。

然后对每个OTUs进行reads数目统计。

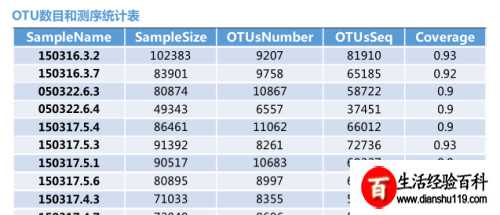
下面2个表其中一个表是对每个样本测序数量和OTU数目进行统计并且在表栺中列出了测序覆盖完整度(显示前10个样本)。

另一个表是对每个样本在分类字水平上数量进行统计并且在表栺中列出了在每个分类字水平上物种数目(显示前10个样本)。

可以看到绝大部分OTU都分类到了属(Genus)也有很多分类到了种(Species)。但是仍然有很多无法完全分类到种一级这是由于环境微生物本身存在非常丰富多样性还有大量菌仍然没有被测序和发现。
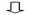
测序数目统计表主要是对每个样本测序数量和OTU数目进行统计并且在表格中列出了测序覆盖完整度(显示前10个样本如果样本超过10个请查看结果中otu_stat.txt文件)
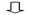
其中 SampleName表示样本名称;SampleSize表示样本序列总数;OTUsNumber表示注释上OTU数目;OTUsSeq表示注释上OTU样本序列总数。
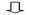
Coverage是指各样品文库覆盖率其数值越高则样本中序列没有被测出概率越低。该指数实际反映了本次测序结果是否代表样本真实情况。

计算公式为:C=1-n1/N 其中n1 = 只含有一条序列OTU数目; N = 抽样中出现总序列数目。

分类水平统计表主要是对每个样本在分类学水平上数量进行统计并且在表格中列出了在每个分类学水平上物种数目(只显示前10个样本如果样本超过10个请查看结果中taxon_all.txt文件)

其中SampleName表示样本名称;Phylum表示分类到门OTU数量;Class表示分类到纲OTU数量;Order表示分类到目OTU数量;Family表示分类到科OTU数量;Genus表示分类到属OTU数量;Species表示分类到种OTU数量。


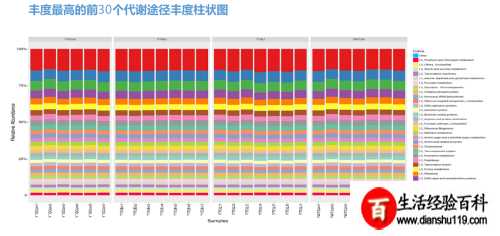
 我们还可以对这些种属构成进行柱状图显示:
我们还可以对这些种属构成进行柱状图显示:
横坐标中每一个条形图代表一个样本纵坐标代表该分类层级序列数目或比例。同一种颜色代表相同分类级别。图中每根柱子中颜色表示该样本在不同级别(门、纲、目等)序列数目序列数目只计算级别最低分类例如在属中计算过了则在科中则不重复计算。

Q: 为什么要选择V3-V4区测序长度?为什么有些文献是V6区有什么区别?

A: 16S rRNA总长约1540 bp包含 9个可变区。由于高通量测序测序长度限制不可能将16S rRNA9个可变区全部测序所以在PCR扩增时往往只能选择1-3个可变区作为扩增片段。Kozich 等评估了Miseq测序仪分析不同16S rRNA可变区准确性发现测定 V4 区效果最佳。根据我们测序长度v3-v4区是最佳选择。



 我们还需要对样本之间或分组之间OTU进行比较获得韦恩图:
我们还需要对样本之间或分组之间OTU进行比较获得韦恩图: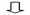
注意韦恩图目前一般最多只能显示5个样本或分组过多样本无法无法进行韦恩图绘制


 样品构成丰度:
样品构成丰度:
稀释曲线
微生物多样性分析中需要验证测序数据量是否足以反映样品中物种多样性稀释曲线(丰富度曲线)可以用来检验这一指标。
稀释曲线是用来评价测序量是否足以覆盖所有类群并间接反映样品中物种丰富程度。稀释曲线是利用已测得16S rDNA序列中已知各种OTU相对比例来计算抽取n个(n小于测得reads序列总数)reads时出现OTU数量期望值然后根据一组n值(一般为一组小于总序列数等差数列)与其相对应OTU数量期望值做出曲线来。当曲线趋于平缓或者达到平台期时也就可以认为测序深度已经基本覆盖到样品中所有物种;反之则表示样品中物种多样性较高还存在较多未被测序检测到物种。
下图中稀释曲线
横坐标代表随机抽取序列数量;纵坐标代表观测到OTU数量。样本曲线延伸终点横坐标位置为该样本测序数量如果曲线趋于平坦表明测序已趋于饱和增加测序数据无法再找到更多OTU;反之表明不饱和增加数据量可以发现更多OTU。

 Shannon-Winner曲线
Shannon-Winner曲线Shannon-Wiener 曲线是利用shannon指数来进行绘制反映样品中微生物多样性指数利用各样品测序量在不同测序深度时微生物多样性指数构建曲线以此反映各样本在不同测序数量时微生物多样性。 当曲线趋向平坦时说明测序数据量足够大可以反映样品中绝大多数微生物物种信息。
与上图一样横坐标代表随机抽取序列数量;纵坐标代表是反映物种多样性Shannon指数。
样本曲线延伸终点横坐标位置为该样本测序数量如果曲线趋于平坦表明测序已趋于饱和增加测序数据无法再找到更多OTU;反之表明不饱和增加数据量可以发现更多OTU。
其中曲线最高点也就是该样本Shannon指数指数越高表明样品物种多样性越高。
Q: Shannon指数怎么算?
A: Shannon指数公式:
其中Sobs= 实际测量出OTU数目;ni= 含有i 条序列OTU数目;N = 所有序列数。


 Rank-Abundance曲线
Rank-Abundance曲线用于同时解释样品多样性两个方面即样品所含物种丰富程度和均匀程度。
物种丰富程度由曲线在横轴上长度来反映曲线越宽表示物种组成越丰富;
物种组成均匀程度由曲线形状来反映曲线越平坦表示物种组成均匀程度越高。
一般超过20个样本图就会变得非常复杂而且不美观所以一般20个样本以下会做该图图片保存为结果目录中rank.pdf。
横坐标代表物种排序数量;纵坐标代表观测到相对丰度。
样本曲线延伸终点横坐标位置为该样本物种数量如果曲线越平滑下降表明样本物种多样性越高而曲线快速陡然下降表明样本中优势菌群所占比例很高多样性较低。

 Alpha多样性(样本内多样性)
Alpha多样性(样本内多样性)Alpha多样性是指一个特定区域或者生态系统内多样性常用度量指标有Chao1 丰富度估计量(Chao1 richness estimator) 、香农 - 威纳多样性指数(Shannon-wiener diversity index)、辛普森多样性指数(Simpson diversity index)等。
计算菌群丰度:Chao、ace;
计算菌群多样性:Shannon、Simpson。
Simpson指数值越大说明群落多样性越高;Shannon指数越大说明群落多样性越高。表中显示前10个样本如果样本大于10个详见结果目录中alpha_div.txt。
Q: 能不能解释下每个指数(如chao1、shannon)?
A: Chao1:是用chao1 算法估计群落中含OTU 数目指数chao1 在生态学中常用来估计物种总数由Chao (1984) 最早提出。Chao1值越大代表物种总数越多。
Schao1=Sobs+n1(n1-1)/2(n2+1)
其中Schao1为估计OTU数Sobs为观测到OTU数n1为只有一条序列OTU数目n2为只有两条序列OTU数目。
Shannon:用来估算样品中微生物多样性指数之一。它与 Simpson 多样性指数均为常用反映 alpha 多样性指数。Shannon值越大说明群落多样性越高。
Ace:用来估计群落中含有OTU 数目指数由Chao 提出是生态学中估计物种总数常用指数之一与Chao1 算法不同。
Simpson:用来估算样品中微生物多样性指数之一由Edward Hugh Simpson ( 1949) 提出在生态学中常用来定量描述一个区域生物多样性。Simpson 指数值越大说明群落多样性越高。
辛普森多样性指数=随机取样两个个体属于不同种概率
=1-随机取样两个个体属于同种概率

 Alpha多样性指数差异箱形图
Alpha多样性指数差异箱形图分别对 Alpha diversity 各个指数进行秩和检验分析(若两组样品比较则使用 R 中wilcox.test 函数若两组以上样品比较则使用 R 中 kruskal.test 函数)通过秩和检验筛选不同条件下显著差异 Alpha Diversity指数。

 Beta多样性分析(样品间差异分析)
Beta多样性分析(样品间差异分析)Beta多样性度量时空尺度上物种组成变化, 是生物多样性重要组成部分, 与许多生态学和进化生物学问题密切相关, 因此在最近10年间成为生物多样性研究热点问题之一。
 PCoA分析
PCoA分析PCoA(principal co-ordinates analysis)是一种研究数据相似性或差异性可视化方法通过一系列特征值和特征向量进行排序后选择主要排在前几位特征值PCoA 可以找到距离矩阵中最主要坐标结果是数据矩阵一个旋转它没有改变样品点之间相互位置关系只是改变了坐标系统。通过PCoA 可以观察个体或群体间差异。
每一个点代表一个样本相同颜色点来自同一个分组两点之间距离越近表明两者群落构成差异越小。PCoA有多张图分别代表PCoA1-2,2-3,3-1。

 NMDS分析(非度量多维尺度分析)
NMDS分析(非度量多维尺度分析)NMDS(Nonmetric Multidimensional Scaling)常用于比对样本组之间差异可以基于进化关系或数量距离矩阵。
横轴和纵轴:表示基于进化或者数量距离矩阵数值 在二维表中成图。
与PCA分析主要差异在于考量了进化上信息。
每一个点代表一个样本相同颜色点来自同一个分组两点之间距离越近表明两者群落构成差异越小。

 PCA分析
PCA分析主成分分析PCA(Principal component analysis)是一种研究数据相似性或差异性可视化方法通过一系列特征值和特征向量进行排序后选择主要前几位特征值采取降维思想PCA 可以找到距离矩阵中最主要坐标结果是数据矩阵一个旋转它没有改变样品点之间相互位置关系只是改变了坐标系统。详细关于主成分分析解释推荐大家看一篇文章http://blog.csdn.net/aywhehe/article/details/5736659 。通过PCA 可以观察个体或群体间差异。
每一个点代表一个样本相同颜色点来自同一个分组两点之间距离越近表明两者群落构成差异越小。
以上三个图可能遇到问题:
1:PCAPcoANMDS分析分别是基于什么数据画?
回答:PCAPcoANMDS分析均是基于OTU分类taxon数据所画用是R语言Vegan包中相关函数画成其中PcoA与NMDS还要基于样本之间距离矩阵才能画成。
2:PCA分析如果图中大部分点集中在一起少数点在很远外围是什么原因造成?
回答:是因为样本OTU分类时候少数样本某些菌含量特别高所造成导致这些样本偏离正常范围建议单独拿出这些样本观察看是否是实验错误。
3:PCA分析时不是有PC1PC2PC3三个坐标吗?是给出三张图吗?还是三维立体图?
回答:PCA作图时会得出PC1PC2PC3三个坐标可以根据PC12,PC13,PC23分别作图一般给出是PC12图当PC12图质量不好看不出明显样本分类效果时可以看PC13或PC23图分类是否清晰也可以用R语言rgl包做出PC123三维图。
QIIME本身结果中有提供PCA三维图结果可以通过网页打开。

 PCAPcoANMDS分析都属于排序分析(Ordination analysis)。排序(ordination)过程就是在一个可视化低维空间或平面重新排列这些样本,使得样本之间距离最大程度地反映出平面散点图内样本之间关系信息。
PCAPcoANMDS分析都属于排序分析(Ordination analysis)。排序(ordination)过程就是在一个可视化低维空间或平面重新排列这些样本,使得样本之间距离最大程度地反映出平面散点图内样本之间关系信息。1、只使用物种组成数据排序称作非限制性排序(unconstrained ordination)(1)主成分分析(principal components analysis,PCA)(2)对应分析(correspondence analysis, CA)(3)去趋势对应分析(Detrended correspondence analysis, DCA)(3)主坐标分析(principal coordinate analysis, PCoA)(4)非度量多维尺度分析(non-metric multi-dimensional scaling, NMDS)
2、同时使用物种和环境因子组成数据排序叫作限制性排序(constrained ordination)(1)冗余分析(redundancy analysis,RDA)(2)典范对应分析(canonical correspondence analysis, CCA)
比较PCA和PCoA:
在非限制性排序中16S和宏基因组数据分析通常用到是PCA分析和PCoA分析两者区别在于:PCA分析是基于原始物种组成矩阵所做排序分析而PCoA分析则是基于由物种组成计算得到距离矩阵得出。在PCoA分析中计算距离矩阵方法有很多种包括如:Euclidean, Bray-Curtis, and Jaccard以及(un)weighted Unifrac (利用各样品序列间进化信息来计算样品间距离其中weighted考虑物种丰度unweighted没有对物种丰度进行加权处理)。
 LDA差异贡献分析
LDA差异贡献分析PCA和LDA差别在于PCA它所作只是将整组数据整体映射到最方便表示这组数据坐标轴上映射时没有利用任何数据内部分类信息是无监督而LDA是由监督增加了种属之间信息关系后结合显著性差异标准测试(克鲁斯卡尔-沃利斯检验和两两Wilcoxon测试)和线性判别分析方法进行特征选择。除了可以检测重要特征他还可以根据效应值进行功能特性排序这些功能特性可以解释顶部大部分生物学差异。详细说明可以参考这篇文章http://blog.csdn.net/sunmenggmail/article/details/8071502 。
不同颜色代表不同样本或组之间显著差异物种。使用LefSe软件分析获得其中显著差异logarithmic LDA score设为2。
问题:LDA分析有什么用?
回答:组间差异显著物种又可以称作生物标记物(biomarkers)该分析主要是想找到组间在丰度上有显著差异物种。

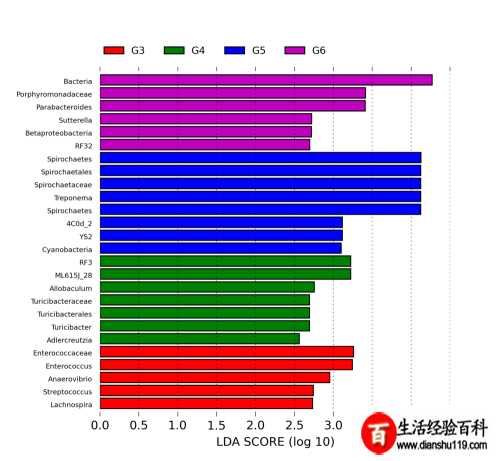
 物种进化树样本群落分布图
物种进化树样本群落分布图是将不同样本群落构成及分布以物种分类树形式在一个环图中展示。数据经过分析后将物种分类树和分类丰度信息通过软件GraPhlAn(http://huttenhower.sph.harvard.edu/GraPhlAn )进行绘制。其目是将物种之间进化关系以及不同样本物种分布丰度和最高分布样本信息在一个视觉集中环图中一次展示其提供信息量较其他图最为丰富。
中间为物种进化分类树不同颜色分支代表不同纲(具体代表颜色见右上角图例)之后外圈灰色标示字母环表示是本次研究中比例最高15个科(字母代表科参见左上角图例)。之后外圈提供是热力图如果样本数<=10个则绘制样本如果样本数超过10个则按照分组绘制每一环为一个样本根据其丰度绘制热力图。最外圈为柱状图绘制是该属所占比例最高样本丰度和样本颜色(样本颜色见环最下方样本名字颜色)。其中热力图和柱状图取值均为原比例值x10000后进行log2转换后值
参考文献:
1. Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. 2013. Emperor: A tool for visualizing high-throughput microbial community data. Gigascience 2(1):16.
2. Legendre, P. and Legendre, L. 1998. Numerical Ecology. Second English Edition. Developments in Environmental Modelling 20. Elsevier, Amsterdam.
3. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation[J]. Genome Biol, 2011, 12(6): R60.
4. Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31: 814–821.

 物种相关性分析
物种相关性分析根据各个物种在各个样品中丰度以及变化情况计算物种之间相关性包括正相关和负相关。
相关性分析使用 CCREPE 算法首先对原始 16s 测序数据种属数量进行标准化然后进行 Spearman 和 Pearson 秩相关分析并进行统计检验计算出各个物种之间相关性之后在所有物种中根据 simscore 绝对值大小挑选出相关性最高前 100 组数据基于 Cytoscap 绘制共表达分析网络图网络图采用两种不同形式表现出来。
物种相关性网络图A:图中每一个点代表一个物种存在相关性物种用连线连接其中红色连线代表负相关绿色先代表正相关连线颜色深浅代表相关性高低。
物种相关性网络图B:图中每一个点代表一个物种点大小表示与其他物种关联关系多少其中与之有相关性物种数越多点半径和字体越大连线粗细代表两物种之间相关性大小连线越粗相关性越高。
参考文献:
Schwager E, Weingart G, Bielski C, et al. CCREPE: Compositionality Corrected by Permutation and Renormalization[J]. 2014.


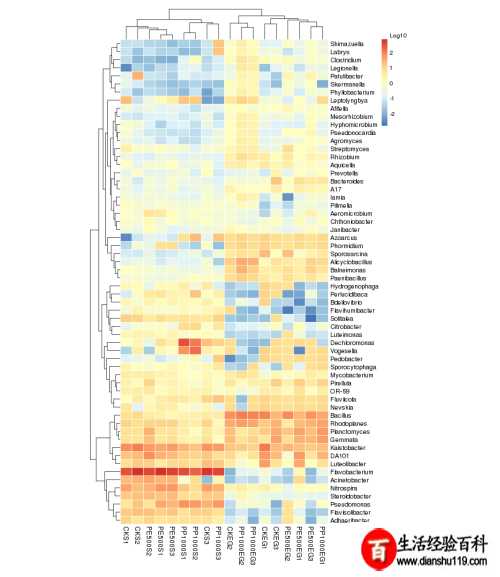
 聚类分析
聚类分析根据OUT数据进行标准化处理(1wlog10)之后选取数目最多前60个物种基于R heatmap进行作图热图中每一个色块代表一个样品一个属丰度样品横向排列属纵向排列两个热图差异是是否对样品进行聚类从聚类中可以了解样品之间相似性以及属水平上群落构成相似性。
如果聚类结果中出现大面积白或黑是因为大量菌含量非常低导致都没有数值可以在绘制之前进行标准化操作对每一类菌单独自身进行Z标准化。

 组间菌群比较选取物种标志物
组间菌群比较选取物种标志物RDA分析
CCA/RDA分析基于对应分析发展一种排序方法将对应分析与多元回归分析相结合每一步计算均与环境因子进行回归又称多元直接梯度分析。主要用来反映菌群与环境因子之间关系。
RDA 是基于线性模型CCA是基于单峰模型。分析可以检测环境因子、样品、菌群三者之间关系或者两两之间关系。
冗余分析可以基于所有样品OTU作图也可以基于样品中优势物种作图;
箭头射线:箭头分别代表不同环境因子;
夹角:环境因子之间夹角为锐角时表示两个环境因子之间呈正相关关系钝角时呈负相关关系。
环境因子射线越长说明该影响因子影响程度越大; 不同颜色点表示不同组别样品或者同一组别不同时期样品图中拉丁文代表物种名称可以将关注优势物种也纳入图中; 环境因子数量要少于样本数量同时在分析时需要提供环境因子数据比如 pH值测定温度值等。

 组间菌群比较选取物种标志物
组间菌群比较选取物种标志物(属水平)组间物种差异性箱形图:
组间物种差异性盒形图描述在不同分组之间具有差异显著某一物种做盒形图图中以属水平为例做物种差异性盒形图。
图中不同颜色代表不同分组更直观显示组间物种差异。每一个盒形图代表一个物种图上方是物种名。

 组间菌群比较选取物种标志物
组间菌群比较选取物种标志物Anosim检验
Anosim分析是一种非参数检验用来检验组间差异是否显著大于组内差异从而判断分组是否有意义。(做组间差异比较分析分组内部至少要3个样本;若样本数不够或组间差异不明显则不生成该图)
R-value介于(-11)之间R-value大于0说明组间差异显著。R-value小于0,说明组内差异大于组间差异统计分析可信度用 P-value 表示P< 0.05 表示统计具有显著性。对Anosim分析结果基于两两样本之间距离值排序获得秩(组间为between组内为within)这样任一两两组比较可以获得三个分类数据并进行箱线图展示(若两个箱凹槽互不重叠则表明它们中位数有显著差异)

 组间菌群比较选取物种标志物
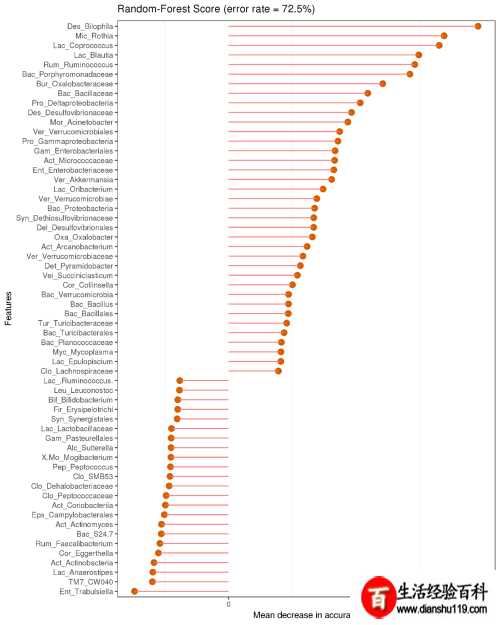
组间菌群比较选取物种标志物随机森林分类树属分类效果
随机森林是机器学习算法一种它可以被看作是一个包含多个决策树分类器。其输出分类结果是由每棵决策树
 投票
投票 结果。由于每棵树在构建过程中都采用了随机变量和随机抽样方法因此随机森林分类结果具有较高准确度并且不需要
结果。由于每棵树在构建过程中都采用了随机变量和随机抽样方法因此随机森林分类结果具有较高准确度并且不需要 减枝
减枝 来减少过拟合现象。随机森林可以有效对分组样品进行分类和预测。
来减少过拟合现象。随机森林可以有效对分组样品进行分类和预测。物种重要性点图。横坐标为重要性水平纵坐标为按照重要性排序后物种名称。上图反映了分类器中对分类效果起主要作用菌属按作用从大到小排列。
Error rate: 表示使用下方特征进行随机森林方法预测分类错误率越高表示基于菌属特征分类准确度不高可能分组之间菌属特征不明显。 图中以所有水平为例取前60个作图。
 组间菌群比较选取物种标志物
组间菌群比较选取物种标志物ROC曲线图
ROC 曲线指受试者工作特征曲线(receiver operating characteristic curve), 是反映敏感性和特异性连续变量综合指标通过构图法揭示敏感性和特异性相互关系。ROC 曲线将连续变量设定出多个不同临界值从而计算出一系列敏感性和特异性再以敏感性为纵坐标、(1-特异性)为横坐标绘制成曲线曲线下面积越大诊断准确性越高。

 组间菌群比较选取物种标志物
组间菌群比较选取物种标志物(属水平)样本-物种丰度关联circos弦装图
样本与物种共线性关系 circus 图是一种描述样本与物种之间对应关系可视化圈图该图不仅反映了每个样本优势物种组成比例同时也反映了各优势物种在不同样本之间分布比例。
样本与物种共线性关系图左半边表示样本属物种丰度情况。右半边表示属水平在不同样本中分布比例情况。在最内一圈:左边不同颜色代表不同物种宽度表示物种丰度圈外数值表示物种丰度刻度值。一端连接右边样本不同颜色代表不同样本条带端点宽度表示该样本中对应物种比例分布。最外两圈:左边不同颜色表示不同样本在某一物种比例右边不同颜色表示不同物种在某一样本中比例。

 组间菌群比较选取物种标志物
组间菌群比较选取物种标志物Ternary三元相图
三元相图是重心图一种它有三个变量在一个等边三角形坐标系中图中某一点位置代表三个变量间比例关系。这里表示三组样本之间优势物种差异通过三元图可以展示出不同物种在分组中比重关系。
三角分别代表三个或三组样本图中圆分别代表排名最高哦属水平物种三种颜色分别代表三组不同分组优势物种圆圈大小代表物种相对丰度圆圈理哪个顶点接近表示此物种在这个分组中含量较高。该分析仅限三个样本或三组样本之间分析比较。

组间菌群比较选取物种标志物
相关系数图
通过 R 软件 corrplot 包绘制spearman 相关性热图并通过该热图可以发现优势物种/样本之间重要模式与关系。
蓝色系为正相关红色系为负相关×表示检验水平下无意义。越靠近颜色条两头相关系数越大。所以说我们可以通过实心圆颜色和大小判断相关方向和相关系数大小。

组间菌群比较选取物种标志物
GraPhlan 图
物种进化树样本群落分布图 GraPhlan 图是将不同样本群落结构及分布以物种分类树形式在一个环图中展示。
使用 GraPhlan 结合 OTU Table对一个分组所有样本 OTU 物种注释结果进行总体展示便于看出优势菌种。
其目是将物种之间进化关系以及不同样本物种分布丰度和最高分布样本信息在一个视觉集中在换图中展示提供信息量较其他图更丰富。
图中中间为物种进化分类树不同颜色分支代表不同纲(具体代表颜色见右上角图例)之后外圈灰色标示字母环表示是本次研究中比例最高 15 个科(字母代表科参见左上角图例)。之后外圈提供是热力图如果样本数 <=10 个则绘制样本如果样本数超过 10 个则按照分组绘制每一环为一个样本根据其丰度绘制热力图。最外圈为柱状图绘制是该属所占比例最高样本丰度和样本颜色(样本颜色见环最下方样本名字颜色)。其中热力图和柱状图取值均为原比例值 x10000后进行 log2 转换后值。

菌群代谢功能预测
通过 16S/ITS 多样性测序可以准确知道群落物种结构但越来越多研究表明微生物群落功能组成比物种组成与环境关系更为密切。基于 16S/ITS 测序结果进行功能预测方法有 PICRUSt、Tax4Fun、FAPROTAX及BugBase
FAPROTAX生态功能预测
FAPROTAX是一款在2016年发表在SCIENCE上较新基于16S测序功能预测软件。它整合了多个已发表可培养菌文章原核功能数据库数据库包含超过4600个物种7600多个功能注释信息这些信息共分为nitraterespiration,methanogenesis, fermentation 和plant pathogenesis等80多个功能分组。
FAPROTAX是基于目前对可培养菌文献资料手动整理原核功能注释数据库其包含了收集自4600多个原核微生物80多个功能分组(如硝酸盐呼吸、产甲烷、发酵、植物病原等)7600多条功能注释信息。
如果PICRUSt在肠道微生物研究更为适合那么FAPROTAX尤其适用于生态环境研究特别是地球化学物质循环分析。FAPROTAX适用于对环境样本(如海洋、湖泊等)生物地球化学循环过程(特别是碳、氢、氮、磷、硫等元素循环)进行功能注释预测。因其基于已发表验证可培养菌文献其预测准确度可能较好但相比于上述PICRUSt和Tax4Fun来说预测覆盖度可能会降低。
参考文献: Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277(2016).
FAPROTAX可根据16S序列分类注释结果对微生物群落功能(特别是生物地化循环相关)进行注释预测。图中横坐标代表样本纵坐标表示包括碳、氢、氮、硫等元素循环相关及其他诸多功能分组。 可快速用于评估样品来源或特征。

基于BugBase表型分类比较
Bugbase也是16年所提供服务一款免费在线16S功能预测工具到今年才发表文章公布其软件原理。该工具主要进行表型预测其中表型类型包括革兰氏阳性、革兰氏阴性、生物膜形成、致病性、移动元件、氧需求包括厌氧菌、好氧菌、兼性菌)及氧化胁迫耐受等7类
参考文献:Thomas A M, Jesus E C, Lopes A, et al. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling[J]. Frontiers in Cellular and Infection Microbiology, 2016, 6.



Picrust群落功能差异分析
通过对已有测序微生物基因组基因功能构成进行分析后我们可以通过16s测序获得物种构成推测样本中功能基因构成从而分析不同样本和分组之间在功能上差异(PICRUSt Nature Biotechnology, 1-10. 8 2013)。
通过对宏基因组测序数据功能分析和对应16s预测功能分析结果比较发现此方法准确性在84%-95%对肠道微生物菌群和土壤菌群功能分析接近95%能非常好反映样品中功能基因构成。
为了能够通过16s测序数据来准确预测出功能构成首先需要对原始16s测序数据种属数量进行标准化因为不同种属菌包含16s拷贝数不相同。然后将16s种属构成信息通过构建好已测序基因组种属功能基因构成表映射获得预测功能结果。(根据属这个水平对不同样本间物种丰度进行显著性差异两两检验我们这里检验方法使用STAMP中two-sample中T-TEST方法Pvalue值过滤为0.05作Extent error bar图。)
此处提供COGKO基因预测以及KEGG代谢途径预测。用户也可自行使用我们提供文件和软件(STAMP)对不同层级以及不同分组之间进行统计分析和制图以及选择不同统计方法和显著性水平。
参考文献:
Donovan H. Parks1 , Gene W. Tyson,STAMP: statistical analysis of taxonomic and functional profiles, Bioinformatics (2014) 30(21): 3123-3124.doi:10.1093
COG构成差异分析图
图中不同颜色代表不同分组列出了COG构成在组间存在显著差异功能分类以及在各组比例此外右侧还给出了差异比例和置信区间以及P-value。

KEGG代谢途径差异分析图
通过KEGG代谢途径预测差异分析我们可以了解到不同分组样品之间在微生物群落功能基因在代谢途径上差异以及变化高低。为我们了解群落样本环境适应变化代谢过程提供一种简便快捷方法。
图解读:图中不同颜色代表不同分组列出了在第三层级构成在组间存在显著差异KEGG代谢途径第三层分类以及在各组比例此外右侧还给出了差异比例和置信区间以及P-value。
本例图所显示是第三层级KEGG代谢途径差异分析也可以针对第二或第一层分级进行分析。

基因差异分析图
除了能对大基因功能分类和代谢途径进行预测外我们还能提供精细功能基因数量和构成预测以及进行样本间以及组间差异分析并给出具有统计意义和置信区间分析结果。
这一分析将我们对于样本群落差异进一步深入到了每一类基因层面。
图解读:图中不同颜色代表不同分组列出了在组间/样本间存在显著差异每一个功能基因(酶)以及在各组比例此外右侧还给出了差异比例和置信区间以及P-value。

在获得标准报告后如果希望单独修改分组或对某些组之间进行显著性差异分析可以使用STAMP软件在自己电脑上进行数据分析。STAMP提供了丰富统计检验方法和图形化结果输出。
在使用STAMP之前需要首先准备需要spf格式文件和样品分组信息表。在我们报告中已经将KEGG和KO以及COG结果文件后经过转换生成了适用于STAMP软件打开spf格式文件还有对应分组信息表文件groupfile.txt。
以下是使用STAMP时一些相关问题详细STAMP使用教程可以参考我们提供STAMP使用教程。
1、 stamp作图用原始数据来源?
STAMP 可以直接使用来自QIIMEbiom文件和PICUSTKEGG和ko 文件groupfile.txt文件格式为tab-saperated value (tab键隔开数据)
2、 分组问题:导入数据之后viewàgroup legend ,在窗口右侧会出现分组栏根据需要进行分组。
3、 Unclassiffied选项中remain Unclassiffied reads、remove Unclassiffied reads、和use only for calculating frequency profiles 方法区别?
remain Unclassiffied reads和use only for calculating frequency profiles方法会保留所有数据而remove Unclassiffied reads仅仅保留有确定分组信息数据。
4、 Statistical test 中Welch’s t-test、t-test、white’s non-parametric t-test区别各自优缺点?
为了确保统计学意义和准确度和精确性需要足够多样本数目t-test检验可以在最少样本数为4时候确保高准确度和精确性。
当两个样本之间具有相同方差时候用t-test更为准确当两个样本没有相同方差Welch’s t-test更为准确。
当样本数目少于8时候可以使用white’s non-parametric t-test该计算时间较长当样本数目过多时候不宜使用该方法。
5、 Two-group 中type: one side 和 two side 区别?
One side 只会显示前一个group与后一个group差异比例而two side 两者之间比例均会显示。
6、 STAMP在使用时首先打开了一个分析文件如果新打开一个可能会导致显示错误?
目前版本STAMP存在一些小问题一次分析只能使用一个数据文件如果要打开新需要关闭软件后再打开。
有其他问题可以联系谷禾信息。